Disease overview
Hypophosphatasia (HPP)
Rare inherited metabolic disease with broad skeletal and dental impact
Overview
Hypophosphatasia is a rare metabolic bone disease caused by loss-of-function mutations in ALPL, the gene encoding tissue non-specific alkaline phosphatase (TNSALP). TNSALP is the key extracellular enzyme that degrades pyrophosphate to allow hydroxyapatite formation and normal bone mineralization. When TNSALP is deficient, pyrophosphate accumulates in the extracellular space, inhibiting hydroxyapatite crystal propagation and causing rickets, osteomalacia, and dental disease. The clinical spectrum is extraordinarily broad, from life-threatening perinatal forms with autosomal recessive inheritance to milder adult-onset disease typically reflecting autosomal dominant mutations with dominant-negative effects. The earlier disease becomes symptomatic, the worse the outcome. Plasma levels of pyrophosphate and pyridoxal 5-phosphate (PLP, the circulating form of vitamin B6) are characteristically elevated and serve as diagnostic biomarkers.
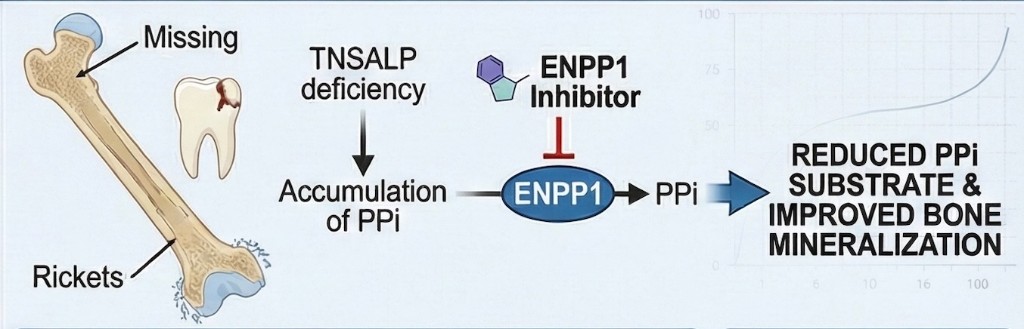
In HPP, TNSALP deficiency leads to accumulation of pyrophosphate. ENPP1 inhibition may reduce PPi substrate and support improved bone mineralization.
Clinical burden
Patients with HPP face bone fragility, recurrent fractures, musculoskeletal pain, premature loss of deciduous teeth, impaired growth and development, and long-term quality-of-life burden. In severe forms, respiratory insufficiency due to a hypomineralized rib cage can be life-threatening. The cost and injection burden of current biological enzyme replacement therapy remain significant challenges, with treatment costs reaching up to $2 million per year and requiring near daily subcutaneous injections.
Current care gap
While enzyme replacement therapy with asfotase alfa has improved outcomes for severe pediatric HPP, the treatment imposes extraordinary cost and injection burden on patients and families. Supportive care including dental management, orthopedic intervention, and pain management remains the standard for milder forms. There is a meaningful need for disease-modifying therapies that address the underlying pyrophosphate imbalance without the limitations of current biological approaches.
Role of ENPP1 in HPP
ENPP1 is the primary enzymatic source of extracellular pyrophosphate, generating PPi from ATP hydrolysis. In HPP, TNSALP deficiency means pyrophosphate cannot be adequately degraded, causing it to accumulate and block skeletal mineralization. Because ENPP1 and TNSALP operate as counter-regulatory enzymes controlling the same pyrophosphate pool, modulating ENPP1 activity represents a mechanistically rational approach to reducing the excess pyrophosphate burden in HPP. Research has shown that correcting the phosphate/pyrophosphate ratio rescues mineralization in HPP periodontal ligament cells (Rodrigues et al., J Periodontol 2011) and that HPP-associated pyrophosphate imbalance disrupts odontoblast gene expression and dentin mineralization (Rodrigues et al., J Endod 2012).
Petragen perspective
Petragen is developing a small molecule approach that targets the ENPP1-mediated overproduction of pyrophosphate, with the goal of restoring the phosphate/pyrophosphate balance needed for normal mineralization. This approach aims to provide a practical, affordable alternative to current biological therapies, relieving the injection burden for patients while bringing costs down dramatically.
References
- Collins MT et al. Skeletal and extraskeletal disorders of biomineralization. Nature Reviews Endocrinology. 2022;18:473-489.
- Rodrigues TL et al. Correction of hypophosphatasia-associated mineralization deficiencies in vitro by phosphate/pyrophosphate modulation in periodontal ligament cells. J Periodontol. 2011;83(7):897-904.
- Rodrigues TL et al. Hypophosphatasia-associated deficiencies in mineralization and gene expression in cultured dental pulp cells obtained from human teeth. J Endod. 2012;38(7):907-912.